
Our research group is interested in the interaction between commensal or pathogenic microorganisms (viruses, bacteria, parasites) and the cells of the gastrointestinal tract and the central nervous system. Particularly, we are interested in the impact of the intestinal microbiota and microbial metabolites on innate immune cells in the gut, liver, joints and CNS. We anticipate that our results will elucidate novel disease mechanisms, will help to identify novel biomarkers and therapeutic options for patients suffering from immune-mediated inflammatory diseases.
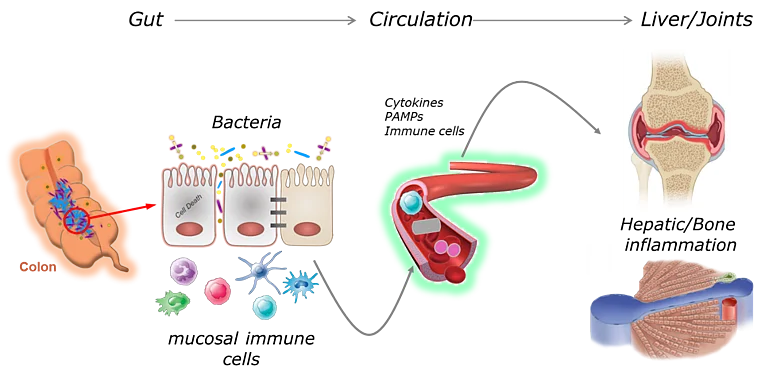
Gut-Liver Axis in Immune-mediated inflammatory diseases
Failure of gut homeostasis as a typical feature of inflammatory bowel disease (IBD) is an important factor in the pathogenesis and progression of systemic inflammation, which can culminate in multiple organ involvement and damage. Up to 30% of IBD patients show biochemical signs for liver injury and hepatobiliary diseases such as primary sclerosing cholangitis (PSC) and autoimmune hepatitis (AIH) are relatively common in IBD. Enteric dysbiosis and translocation of bacteria across the gut epithelial barrier have been widely recognized as major factors in the progression of chronic liver disease by promoting hepatocellular injury and inflammation. However, the sequence of events and the underlying molecular mechanisms are poorly defined. Recent studies by our group have revealed important functions for programmed necrosis in the pathogenesis of gastrointestinal and hepatic inflammation and implicated that programmed necrosis could be implicated in the pathogenesis of many human inflammatory diseases. The proposed project aims at a multidisciplinary approach to characterize the association between programmed necrosis in the gut and the initiation/progression of hepatic inflammation. This comprehensive project will advance our understanding of mechanisms linking failure of gut homeostasis to hepatic inflammation by replacing the organ centered point of view by an interdisciplinary approach that includes analysis in both affected organs (liver and gut). This will provide the basis for the development of a more efficient and safer therapy for IBD patients with clinical/biochemical indications for hepatobiliary involvement.
Gut-Joint Axis in Immune-mediated inflammatory diseases
Intestinal inflammation and dysbiosis have been linked to autoimmune diseases such as rheumatoid arthritis (RA), however, the underlying mechanisms remain incompletely understood. Our own published and preliminary data suggest that expression of caspase-8 and hypoxia-inducing factors (HIF) have a crucial influence on mucosal immunity and the composition of the intestinal microbiota. Further preliminary data suggest that both factors might additionally control systemic Th17-driven (auto-) immunity and the onset of arthritis. The proposed project aims at a multidisciplinary approach to characterize the link between intestinal epithelial cell (IEC) death and the initiation/progression of joint inflammation. In particular, we aim to dissect host-microbial interaction and determine the influence of this interaction (epithelium-microbiota) on systemic immunity and the onset of autoimmune arthritis.