
Arbeitsgruppe Prof. Dr. rer. nat. Claudia Günther
Mukosale Infektionsbiologie
Unsere Forschungsgruppe interessiert sich für die Interaktion zwischen kommensalen oder pathogenen Mikroorganismen (Viren, Bakterien, Parasiten) und den Zellen des Magen-Darm-Trakts sowie des zentralen Nervensystems.
Insbesondere für die Auswirkungen der intestinalen Mikrobiota und mikrobieller Metaboliten auf angeborene Immunzellen im Darm, in der Leber, in den Gelenken und im ZNS.
Wir erwarten, dass unsere Ergebnisse neue Krankheitsmechanismen aufklären und dazu beitragen werden, neue Biomarker und therapeutische Optionen für Patienten mit immunvermittelten Entzündungskrankheiten zu identifizieren.
Projekte
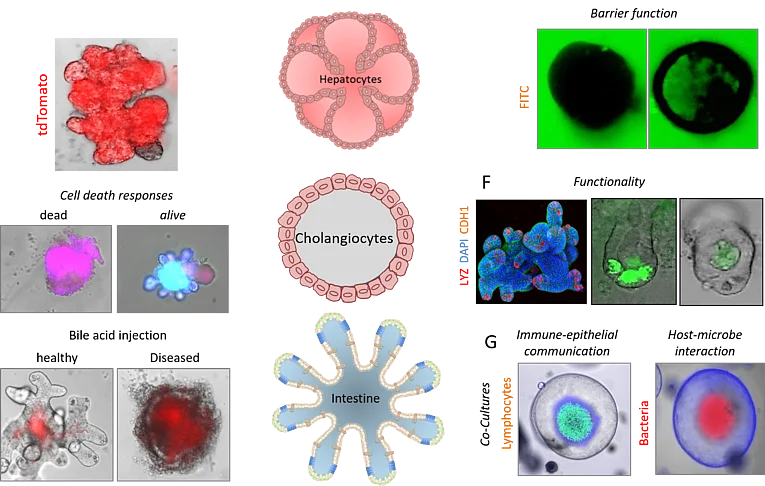
Ex vivo organ cultures Ex vivo organ cultures
Organoids are three-dimensional organ-like structures that can be generated from organ-specific, embryonic or pluripotent stem cells. Organoids can recapitulate physiologically relevant organ-like properties and are therefore of particular importance for medical research. Over the past 9 years, we have successfully worked with organoids and have been able to establish organoid cultures from various mouse tissues such as the liver, gallbladder, pancreas, stomach, and intestine. In addition, we have developed a variety of technologies including genome editing, transfection, cryopreservation, microinjection, co-culture, and a wide range of functional assays. We use organoids to study the molecular and cellular mechanisms of various diseases, to develop new drugs, and for clinical applications in regenerative therapy and personalized medicine.
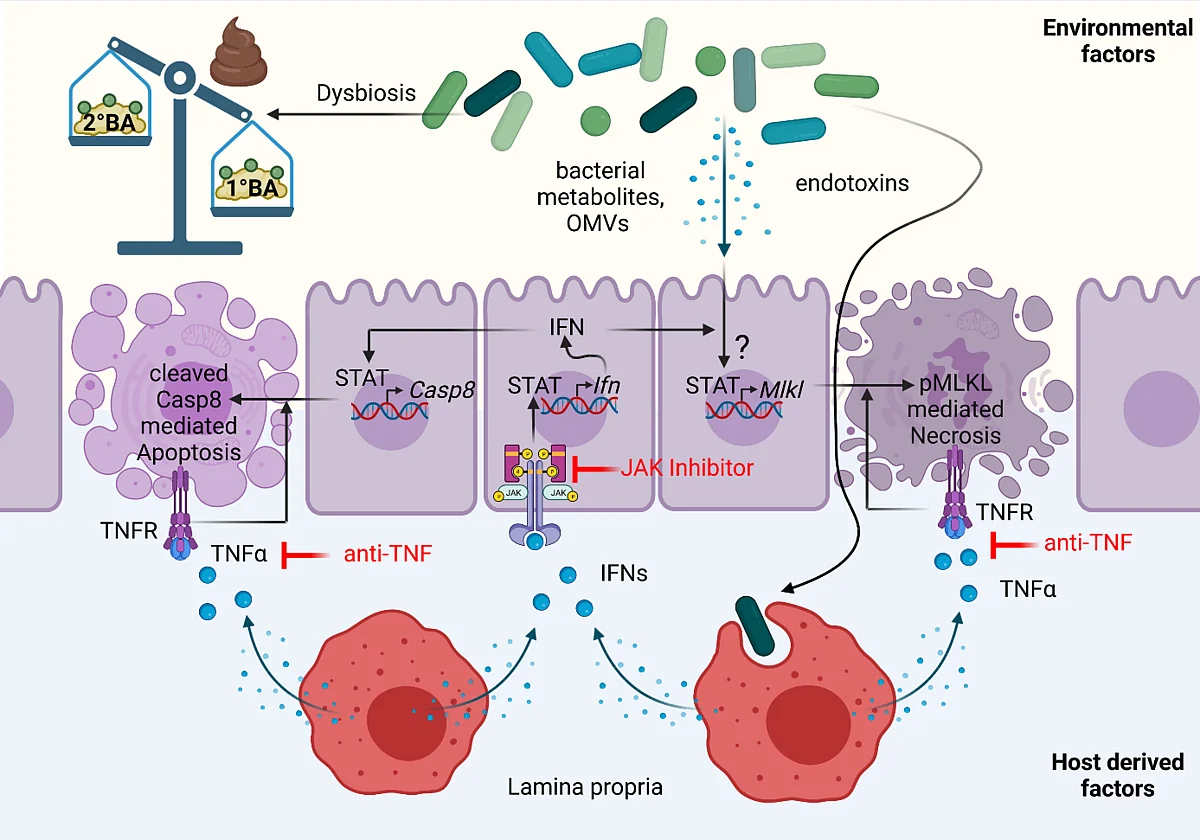
TRR241 "Immune epithelial signaling pathways in inflammatory bowel disease."
Mechanisms linking IFN-mediated cell death with bile acid signaling in small intestinal inflammation (A02)
In previous work, we uncovered that mucosal barrier dysfunction caused by regulated necrosis was strongly linked to microbial dysbiosis and chronic intestinal inflammation both in preclinical mouse models and human inflammatory bowel disease (IBD) patients. In the current funding period, we not only identified Interferon (IFN) signaling as a strong inducer of regulated necrosis in the gut, but in collaboration with several TRR241 projects further demonstrated that in the context of Crohn´s disease (CD), TNFα, and IFNs act synergistically to trigger Paneth cell death and barrier dysfunction by Mixed lineage kinase domain-like protein (MLKL)-dependent regulated necrosis (necroptosis). Based on these collaborative results, we proposed a novel model in which regulated necrosis, in genetically predisposed individuals, plays a central role during the onset of inflammation and in general results in the perpetuation of inflammation. Furthermore, we could show that genetic susceptibility alone is sufficient to drive mild inflammation but that environmental factors such as a disease-relevant microbial flora, determine the extent and localization of inflammation in the gut. Our observation that a distinct, not yet identified, population in the mucosal microbiota is able to transcriptionally influence cell death mechanisms in the intestinal epithelium, confirmed the current understanding of IBD, that not only host-dependent mechanisms, but also the microenvironment, are critical factors that can promote immune-mediated inflammation. Preliminary data of the ongoing funding period, further indicate that IFN-induced MLKL-mediated Paneth cell death is controlled by microbiota-dependent and -independent Bile Acid (BA) signaling. Accordingly, we observed an increased BA signaling in the context of small intestinal inflammation (Casp8ΔIEC mouse model and CD patient cohort) particularly in the terminal ileum, the main location of inflammation in this prototype of IBD. Of note, by taking advantage of a preclinical mouse model for CD (Casp8ΔIEC model), we uncovered that this effect was strongly dependent on environmental factors, since enhanced BA signaling was absent under germ-free conditions. Supported by this TRR, we further uncovered that primary BAs (1°BAs) trigger epithelial cell, while secondary BAs (2°BAs) negatively regulate Mlkl gene expression and thus might directly inhibit necroptosis. As we found that BA further influence T cell function and elasticity, BA might also indirectly affect inflammatory epithelial cell death. By taking advantage of the IBDome cohort, we further demonstrated that key signaling molecules of BA homeostasis are differentially expressed between IBD prototypes. These data implicate a mechanistic link between enhanced IFN signaling, microbial dysbiosis and Paneth cell death that is regulated by BA signaling. In the second funding period, we will therefore take advantage of the unique expertise within the TRR 241 to investigate the impact of BA signaling on IFN-mediated cell death and its implication for immune-epithelial communication and inflammation. The long-term vision of this project is to identify how environmental factors shape inflammatory epithelial cell death and barrier dysfunction directly or indirectly via immune cells. We are confident that these studies uncover novel mechanisms that can be targeted for therapeutic approaches.
Further information: https://www.transregio241.de/
TRR305 “Striking a moving target: From mechanisms of metastatic organ colonisation to novel systemic therapies”
Angiocrine signaling in metastatic colonization of colon cancer (B08)
Colorectal cancer (CRC) is among the major death-causing cancers worldwide and distant metastases are the main cause of cancer-related death in the respective patients. The mutual interaction of disseminated cancer cells (DCCs) with blood vessel endothelial cells (ECs) is involved in metastatic colonization of hostile tissues, but it is not known whether it may foster or prevent the growth of DCCs. We previously uncovered that tumor blood vessels, in contrast to the current knowledge, not only promote but under certain conditions also suppress tumorigenesis. We found that this function requires SPARCL1, a matricellular protein which was highly associated with quiescent ECs in mature vessels of normal tissues, including colon, stomach and lung. Of note, SPARCL1 is released from quiescent ECs by classical secretion and therefore acts as a para- and autocrine mediator. Functional studies demonstrated that SPARCL1 regulates blood vessel homeostasis by inhibiting EC proliferation and migration. Interestingly, high SPARCL1 expression was observed in CRC tissues with a prognostically favorable Th1-like tumor immune microenvironment (Th1-TME) and reduced angiogenic activity. In contrast, SPARCL1 expression was downregulated and low in aggressive CRCs devoid of a Th1 immune response but with high angiogenic activity. On a functional level, SPARCL1 inhibited proliferation and migration of endothelial cells and CRC tumor cells. These findings indicated that SPARCL1 represents a novel angiocrine suppressor of neo-angiogenesis and tumor progression. Accordingly, the central hypothesis of this proposal is that angiocrine SPARCL1 inhibits expansion of single DCCs residing in distant organs and by this mechanism actively counteracts loco-regional and distant metastasis. Within this project, we will analyze whether and how SPARCL1 actively counteracts metastatic colonization in distant organs. The long-term perspective is to establish SPARCL1 as a novel diagnostic marker to predict the risk of metastasis and to discover novel functions of matricellular proteins that could be targeted for future therapeutic intervention of metastasis.
Further information: https://www.trr305.de/
This is a collaborative project with the Department of Experimental Surgery (Prof. Dr. rer. nat. Elisabeth Naschberger)
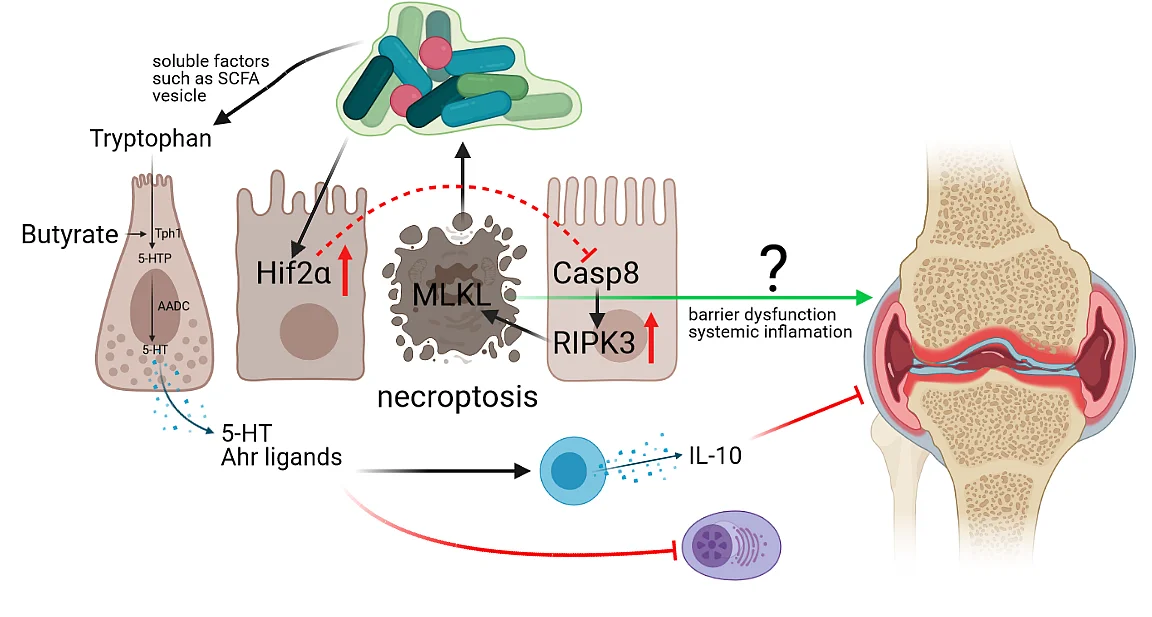
FOR2886 “Pathways triggering Autoimmunity and Defining Onset of early Reumatoid Arthritis”
Intestinal epithelial cell death as modulator of intestinal dysbiosis, systemic autoimmunity and the onset of arthritis (A02)
Summary: Intestinal inflammation and dysbiosis have been linked to autoimmune diseases such as rheumatoid arthritis (RA), however, the underlying mechanisms remain incompletely understood. During the first funding period, we have shown that dysregulated intestinal epithelial cell death by altered CASP8 or HIF2α protein function affects the gut microbiota and subsequently immune cell functions locally in the gut and in the periphery. On a functional level, we could further uncover that HIF proteins influence tight junction biology and epithelial cell death upstream of caspase-8 with opposing functions in experimental arthritis. Interestingly, we further identified that epithelial necroptosis influences tryptophan metabolism and that supplementation of SCFAs ameliorates intestinal inflammation in CASP8 deficient mice. These data indicate that targeting intestinal epithelial cell death might represent a novel promising therapeutic option particularly during the early phase of arthritis. During the second funding period, we therefore propose to better delineate the link between epithelial hypoxia induced necroptosis, SCFAs and arthritis.
Further information: https://www.pandora.for2886.forschung.fau.de/
This is a collaborative project with the Department of Rheumatology (Prof. Dr. rer. nat. Aline Bozec)
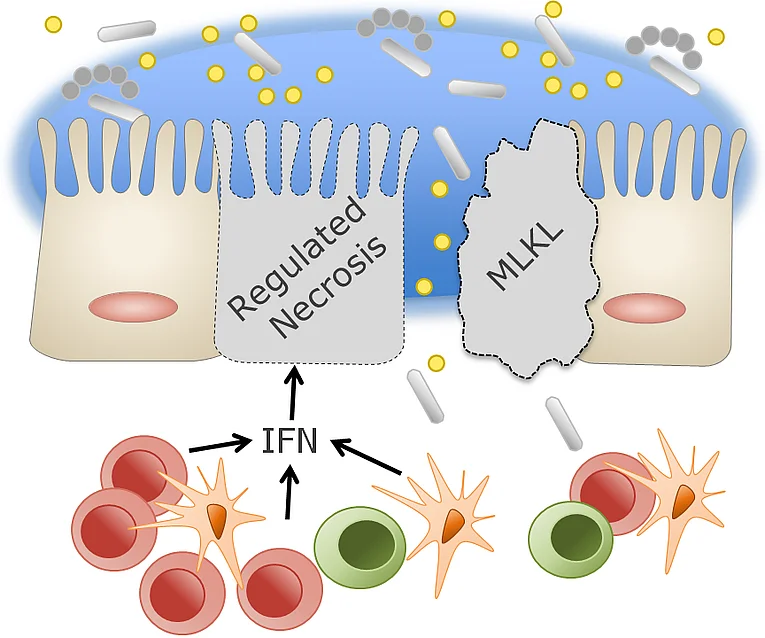
Linking mechanisms between IFN-mediated activation of mixed lineage kinase domain-like protein and intestinal inflammation.
On the one hand, interferons (IFNs) are potent immunomodulatory cytokines expressed mainly by cells of the intestinal epithelium and mucosa in response to viral and bacterial infections. On the other hand, IFNs promote cell death of the intestinal epithelium, thus affecting its tight junction barrier, and are thus involved in the development of inflammatory bowel diseases. Consistent with this, we found a correlation between IFN expression (especially IFN-λ) and disease activity in Crohn's disease patients. In addition, we identified a previously undescribed type of regulated necrosis that is highly dependent on IFN signaling and proceeds via a mixed lineage kinase domain-like protein (MLKL)-dependent signaling pathway, but which is independent of RIPK3 and caspase-8. Because IFN-λ-dependent epithelial cell death is linked to strong upregulation of MLKL, we hypothesize that IFN-λ induces MLKL-dependent programmed necrosis in the intestine, thereby controlling the development of inflammatory bowel disease in mice and human patients. We are addressing the question of whether and through which signaling pathways IFN-regulated necrosis in epithelial cells contributes to the development of inflammatory bowel disease and how this can be therapeutically addressed in the future.

IZKF Jochen Kalden Gruppe (N5)
Role of extracellular vesicles as biological shuttle system for inter-kingdom communication and organ crosstalk
Immune-based therapies and diagnostics have proven their enormous potential in the treatment of immune-mediated inflammatory diseases (IMIDs) such as inflammatory bowel disease (IBD), autoimmune hepatitis (AIH), rheumatoid arthritis (RA), multiple sclerosis (MS) and are currently revolutionizing medical practice across numerous disciplines, allowing for highly individualized therapy approaches. Insights into the underlying molecular disease mechanisms thus become increasingly important for researchers and clinicians alike. Common denominators of such diseases are progressive inflammation-driven tissue damage and consecutive dysfunction of the affected organs. My group has been focusing on how tissue injury and the associated response mechanisms are modulated by the microenvironment with a particular interest on IMIDs of the liver and the gut (Wittkopf & Günther et al., Gastroenterology 2013; Günther et al., Gut 2013; Günther et al., JCI 2016; Bittel et al., Cell Death Dis. 2019). A major strength in this endeavour is represented by our interdisciplinary and translational research approach, spanning the spectrum from basic cellular and molecular methods to pre-clinical models and the validation of our findings in a clinical setting using human material. This strategy has successfully resulted in several major observations with a strong clinical relevance for immune-based therapies and diagnostics.
Further information: https://www.izkf.med.fau.de/nachwuchs/jochen-kalden-foerderprogramm/n5-organ-crosstalks-in-imids-guenther/
DFG Single Grand
Gut‐Liver axis: The interrelated role of regulated necrosis as key driver of gastrointestinal and hepatic inflammation
Failure of gut homeostasis as a typical feature of inflammatory bowel disease (IBD) is an important factor in the pathogenesis and progression of systemic inflammation, which can culminate in multiple organ involvement and damage. Up to 30% of IBD patients show biochemical signs for liver injury and hepatobiliary diseases such as primary sclerosing cholangitis (PSC) and autoimmune hepatitis (AIH) are relatively common in IBD. Enteric dysbiosis and translocation of bacteria across the gut epithelial barrier have been widely recognized as major factors in the progression of chronic liver disease by promoting hepatocellular injury and inflammation. However, the sequence of events and the underlying molecular mechanisms are poorly defined. Recent studies by our group have revealed important functions for programmed necrosis in the pathogenesis of gastrointestinal and hepatic inflammation and implicated that programmed necrosis could be implicated in the pathogenesis of many human inflammatory diseases. The proposed project aims at a multidisciplinary approach to characterize the association between programmed necrosis in the gut and the initiation/progression of hepatic inflammation. This comprehensive project will advance our understanding of mechanisms linking failure of gut homeostasis to hepatic inflammation by replacing the organ centered point of view by an interdisciplinary approach that includes analysis in both affected organs (liver and gut). This will provide the basis for the development of a more efficient and safer therapy for IBD patients with clinical/biochemical indications for hepatobiliary involvement.
Further information: https://www.usz.ch/team/andreas-kremer/
This is a collaborative project with the Universitäts Spital Zürich, Prof. Dr. med. Andreas Kremer
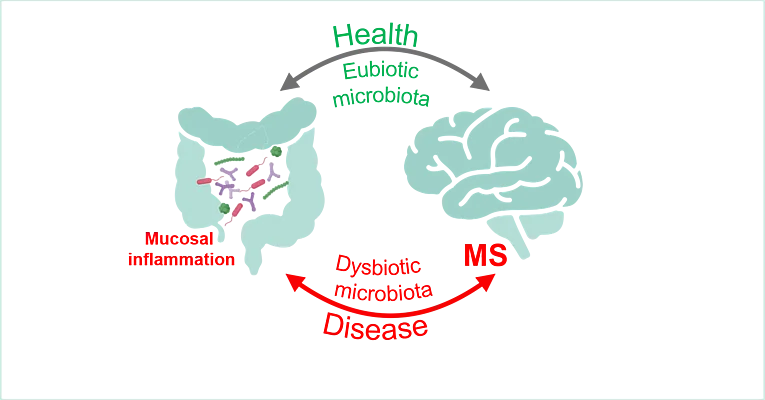
FOR 5024 „Immune checkpoints of Gut-Brain communication in inflammatory & neurodegenerative diseases (GB.com)”
Impact of microbiota-brain communication on MS-related autoimmunity (B01)
Summary: Multiple sclerosis (MS) is the most common chronic inflammatory autoimmune disease of the central nervous system (CNS) and starts in young individuals in the most productive period of their lives. Its multifactorial pathogenesis includes genetic and environmental factors such as communication of the gut microbiota with peripheral and central immune processes. Accordingly, previous studies have demonstrated that commensal microorganism in the gut can modulate immunological processes both in the peripheral immune system as well as within the CNS and alterations in gut microbial composition have been identified as essential component of neuroinflammation. In this context, published and preliminary data from our groups suggest that microbial derived factors participate in the onset and progression of autoimmune CNS inflammation in MS. However, our mechanistic understanding of these processes is still limited. Within this project, we will dissect microbiota-gut-brain communication via bacterial-derived extracellular vesicles and determine how this interaction affects the CNS and MS related autoimmunity. The long-term vision of this project is to identify novel routes of microbiota-brain crosstalk that can be targeted for therapeutic approaches.
This is a collaborative project with the Department of Neurology, Prof. Dr. med. Veit Rothhammer
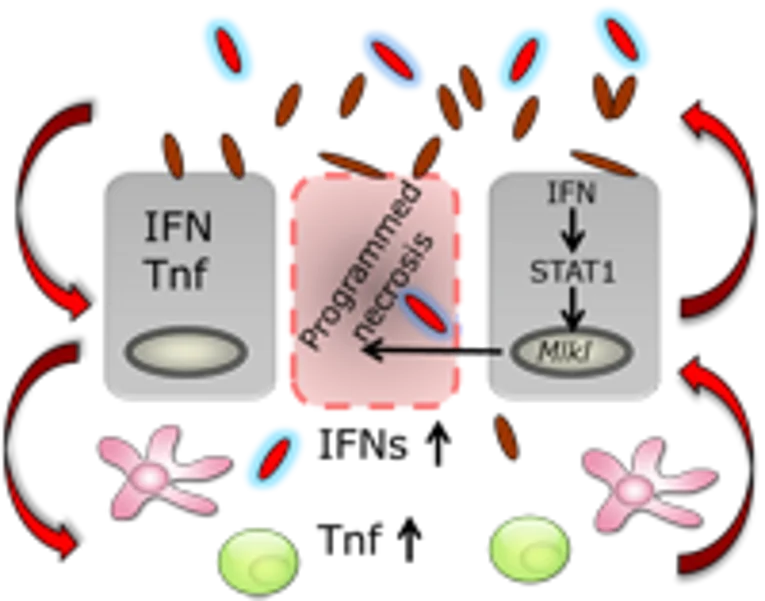
The effects of the intestinal microbiota as well as microbiota-mediated immune responses on the cell death machinery of the intestinal epithelium
Intestinal epithelial cells form a physical barrier to separate commensal and pathogenic microorganisms from the underlying immune system. Specialized intestinal epithelial cells (e.g. Paneth cells, goblet cells) produce mucus and antimicrobial peptides which impede the access and survival of bacteria. Given these basic and diverse functions of intestinal epithelial cells, it is evident that proliferation, differentiation, and cell death of these cells must be tightly controlled to maintain intestinal homeostasis. Our studies have already identified a crucial role of necroptosis in regulating intestinal homeostasis and intestinal barrier, which strongly contributes to intestinal inflammation. However, little is known about the effects of the intestinal microbiota on this particular form of cell death and the effects of epithelial necroptosis on the intestinal flora. The goal of this project is to define the bidirectional influence of the intestinal microbiota and cell death machinery under physiological and pathophysiological conditions. A better understanding of host-microbe interactions related to the establishment and maintenance of gut barrier function is essential for the development of new strategies to treat inflammatory bowel disease and gastrointestinal infections.
Intestinal epithelial cell death as a modulator of intestinal microbiota, systemic autoimmunity, and initiator of arthritis
Gastrointestinal inflammation and dysbiosis are associated with autoimmune diseases such as rheumatoid arthritis (RA). However, the underlying molecular mechanisms are incompletely understood and elucidated. Our preliminary data suggest that the expression of cell death regulators, such as caspase-8, have a critical impact on mucosal immune mechanisms and the composition of the intestinal microbiota. Further preliminary data suggest that caspase-8 may additionally influence systemic Th17-driven (auto)immunity and the occurrence of arthritis. Therefore, in a multidisciplinary approach, we would like to investigate the relationship between cell death of the intestinal epithelium and the development of joint inflammation.
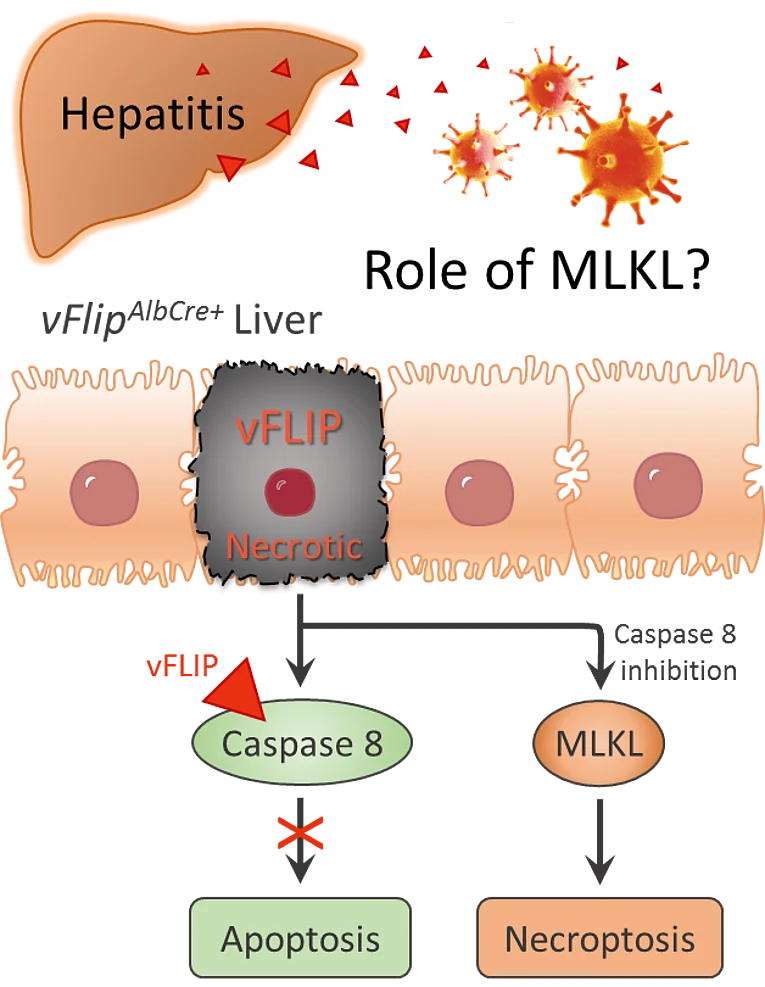
Role of MLKL-mediated necrosis in the development of hepatitis
Programmed cell death plays an essential role in almost all liver diseases. A more comprehensive understanding of potential triggers and molecular mechanisms of cell death in the liver is therefore crucial for the development of new therapeutic approaches. In this project, we therefore analyzed the role of mixed-lineage kinase domain-like pseudokinase (MLKL)-mediated necrosis in hepatitis. Indeed, we found that the transcription rate of MLKL is increased in the liver of hepatitis C patients. To further investigate the influence of MLKL in virus-induced hepatitis, we established a new mouse model that transgenically expresses the viral cell death regulator vFLIP (caspase-8 inhibitor) in a liver-specific manner. Excessive production of this viral apoptosis inhibitor caused extensive MLKL-mediated necrosis in the liver, eventually leading to liver failure and early death of the mice.
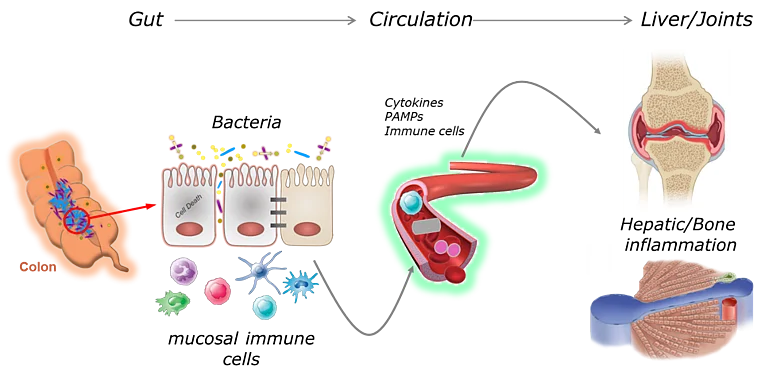
Gut-Liver Axis in Immune-mediated inflammatory diseases
Failure of gut homeostasis as a typical feature of inflammatory bowel disease (IBD) is an important factor in the pathogenesis and progression of systemic inflammation, which can culminate in multiple organ involvement and damage. Up to 30% of IBD patients show biochemical signs for liver injury and hepatobiliary diseases such as primary sclerosing cholangitis (PSC) and autoimmune hepatitis (AIH) are relatively common in IBD. Enteric dysbiosis and translocation of bacteria across the gut epithelial barrier have been widely recognized as major factors in the progression of chronic liver disease by promoting hepatocellular injury and inflammation. However, the sequence of events and the underlying molecular mechanisms are poorly defined. Recent studies by our group have revealed important functions for programmed necrosis in the pathogenesis of gastrointestinal and hepatic inflammation and implicated that programmed necrosis could be implicated in the pathogenesis of many human inflammatory diseases. The proposed project aims at a multidisciplinary approach to characterize the association between programmed necrosis in the gut and the initiation/progression of hepatic inflammation. This comprehensive project will advance our understanding of mechanisms linking failure of gut homeostasis to hepatic inflammation by replacing the organ centered point of view by an interdisciplinary approach that includes analysis in both affected organs (liver and gut). This will provide the basis for the development of a more efficient and safer therapy for IBD patients with clinical/biochemical indications for hepatobiliary involvement.
Gut-Joint Axis in Immune-mediated inflammatory diseases
Intestinal inflammation and dysbiosis have been linked to autoimmune diseases such as rheumatoid arthritis (RA), however, the underlying mechanisms remain incompletely understood. Our own published and preliminary data suggest that expression of caspase-8 and hypoxia-inducing factors (HIF) have a crucial influence on mucosal immunity and the composition of the intestinal microbiota. Further preliminary data suggest that both factors might additionally control systemic Th17-driven (auto-) immunity and the onset of arthritis. The proposed project aims at a multidisciplinary approach to characterize the link between intestinal epithelial cell (IEC) death and the initiation/progression of joint inflammation. In particular, we aim to dissect host-microbial interaction and determine the influence of this interaction (epithelium-microbiota) on systemic immunity and the onset of autoimmune arthritis.

